Overview¶
Preparing input files¶
Before running AMP2, two input files should be prepared such as YAML style configuration file (config.yaml) and structure file. The details for input files are explained in Input files. The basic format of config.yaml and structure files are like below:
config.yaml:
directory: submit: ./Submit # the path of structure file or the directory containg structure files output: ./Output # the path of the directory where calculation is conducted done: ./Done # the path of the directory where results are saved error: ./ERROR # the path of the directory where the materials with error are saved src_path: ./src # the path of the directory of AMP2 source codes pot_path_gga: ./pot/PBE # the path of directory for GGA pseudopotential pot_path_lda: ./pot/LDA # the path of directory for LDA pseudopotential program: vasp_std: ./vasp_std # the path of standard version of VASP vasp_gam: ./vasp_gam # the path of gamma-only version of VASP vasp_ncl: ./vasp_ncl # the path of noncollinear version of VASP gnuplot: /usr/local/bin/gnuplot # the path of executable file for gnuplot mpi_command: mpirun # mpi command (ex. mpirun, mpiexec, ...) vasp_parallel: npar: 2 # the number of bands that are treated in parallel. It is same to NPAR tag in VASP. kpar: 2 # the number of kpoints that are treated in parallel. It is same to NPAR tag in VASP.Structure file (VASP structure file format):
Primitive Cell 1.000000000 0.0 2.714895 2.714895 2.714895 0.0 2.714895 2.714895 2.714895 0.0 Si 2 Selective dynamics Direct 0.5 0.5 0.5 T T T ! Si1 0.75 0.75 0.75 T T T ! Si1
Running AMP2¶
You can execute AMP2 using shell script as following.
sh run.sh
The details for shell script are mentioned in the section, “Execution AMP2” in Installation and execution.
Outputs¶
After starting the calculation, new directory is formed in output_path as the name of the structure file. (name directory is formed from name.cif or POSCAR_name.) Then, if calculation is well finished, the directory moves to done_path. If not, it moves to error_path. The following data are the examples of calculation results for Cr2O3. More details for output files are written in Output.
POSCAR_GGA:
relaxed poscar 1.000000000 2.53085784423 1.46119145764 4.60391533726 -2.53085784423 1.46119145764 4.60391533726 0.0 -2.9223829153 4.60391533726 Cr O 4 6 Selective dynamics Direct 0.348055231569 0.348055231569 0.348055231569 T T T ! Cr1_up 0.848055231569 0.848055231569 0.848055231569 T T T ! Cr1_up 0.151944768431 0.151944768431 0.151944768431 T T T ! Cr1_down 0.651944768431 0.651944768431 0.651944768431 T T T ! Cr1_down 0.553903778143 0.946096221857 0.25 T T T ! O1 0.946096221857 0.25 0.553903778143 T T T ! O1 0.25 0.553903778143 0.946096221857 T T T ! O1 0.0539037781426 0.75 0.446096221857 T T T ! O1 0.75 0.446096221857 0.0539037781426 T T T ! O1 0.446096221857 0.0539037781426 0.75 T T T ! O1Band_gap_GGA.log:
Band gap: 2.734 eV (Indirect) VBM: 0.2916667 0.0 0.0 : 3.366 eV CBM: 0.42206 0.42206 -0.01078659 : 6.100 eV nVBM: 30 spin: 1 nCBM: 31 spin: 1band_GGA.png:

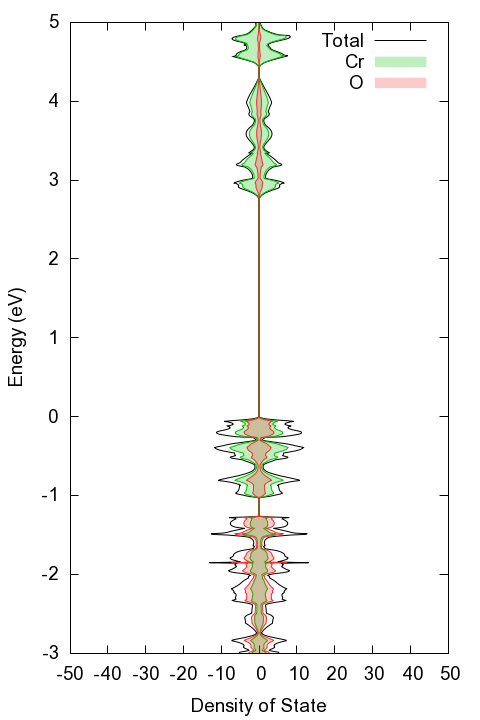
dos_GGA.png:
List of source codes¶
AMP2 consists of several python codes as follows:
- main.py:
- This is main code to run AMP2.
- amp2_input.py:
- This is for generating input files for VASP from structure file.
- kpoint.py:
- This is for conducting a convergence test of k-points.
- cutoff.py:
- This is for conducting a convergence test of cutoff energy.
- relax.py:
- This is for conducting structure optimization.
- magnetic_ordering.py:
- This is for identifying the most stable magnetic spin ordering.
- band.py:
- This is for drawing band structure and estimating band gap.
- dos.py:
- This is for drawing density of states.
- hse_gap.py:
- This is for estimating band gap with PBE@HSE scheme.
- effm.py:
- This is for estimating effective masses of hole and electron.
- dielectric.py:
- This is for estimating dielectric tensor.
- get_result.py:
- This is for summarizing the calculation results.
- input_conf.py:
- This is for handling YAML type configuration.
- rerun_for_metal.py:
- This is a code to restart the all calculations without the on-site U term if the material was found to be metallic and U was applied.
- genetic_algorithm.py:
- This is for performing genetic algorithm to find the most stable magnetic spin ordering.
- genetic_operator.py:
- This is a package of modules for performing genetic algorithm.
- make_supercell.py:
- This is a code to build supercell to find magnetic primitive cell.
- mk_suprecell.py:
- This is a code to build supercell for the Ising coefficient.
- module_subr.py:
- This is a package of modules for ‘mk_supercell.py’.
- module_amp2_input.py:
- This is a package of modules for generating input files for VASP from structure file.
- module_converge.py:
- This is a package of modules for convergence test.
- module_relax.py:
- This is a package of modules for structure optimization.
- module_AF.py:
- This is a package of modules for identifying the most stable magnetic spin ordering.
- module_GA.py:
- This is a package of modules for genetic algorithm.
- module_band.py:
- This is a package of modules for drawing band structure and calculating band gap.
- module_dos.py:
- This is a package of modules for drawing density of states.
- module_hse.py:
- This is a package of modules for calculating band gap with HSE@PBE scheme.
- module_effm.py:
- This is a package of modules for calculating effective mass.
- module_dielectric.py:
- This is a package of modules for calculating dielectric tensor.
- module_vasprun.py:
- This is a package of modules to run VASP.
- module_log.py:
- This is a package of modules to record log.
- module_vector.py:
- This is a package of modules to calculate several properties such as distance between two points and angle.
Additionally, there are files for predefined variables.
- INCAR0:
- This is for default configuration for ‘INCAR’.
- U_table.yaml:
- This is for default U parameters.
- pot_table.yaml:
- This is for default potential files.
- config_def.yaml:
- This is default configuration for ‘config.yaml’.